General tutorial
This tutorial shows you the full processing pipeline, from generating the necessary session metadata from the template, running the processing pipeline, opening multiple sessions for manual curation in CellExplorer, and finally using the cell_metrics for filtering cells, by two different criteria. The tutorial is also available as a Matlab script: (tutorials/CellExplorer_Tutorial.m).
- Define the basepath of the dataset to run. The dataset should ideally consist of the raw data
basename.dat, and spike sorted data.basepath = '/your/data/path/basename/'; cd(basepath) - Generate session metadata struct using the template function and display the metadata in the session GUI:
session = sessionTemplate(basepath,'showGUI',true);
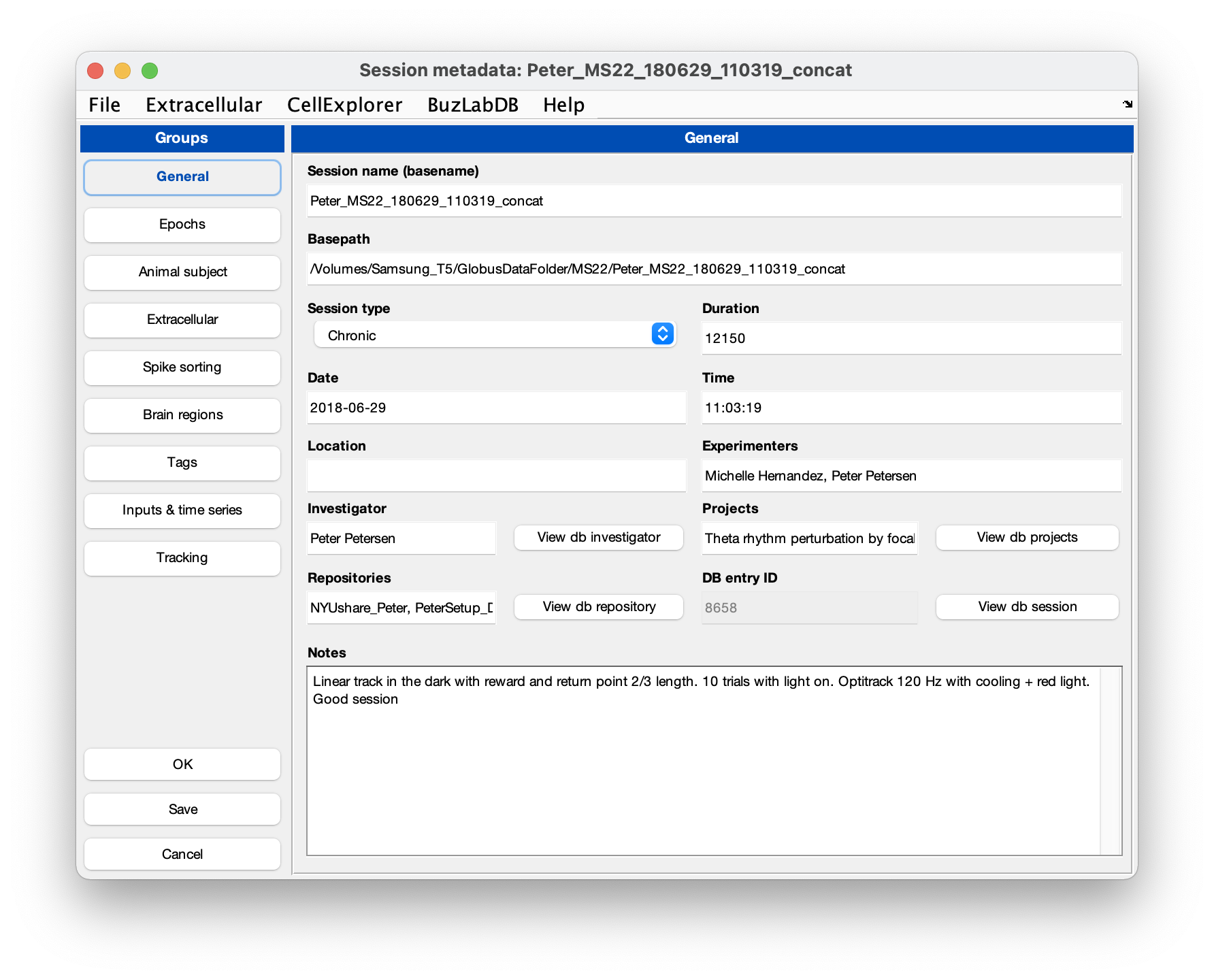
You can use the GUI to inspect the metadata. Make sure that the extracellular tab is filled out correctly for your data (see screenshot below). The template script can extracted existing metadata from a Neuroscope compatible basename.xml, from Intan’s info.rhd file, from KiloSort’s rez.mat file, and from a basename.sessionInfo.mat (Buzcode) file.
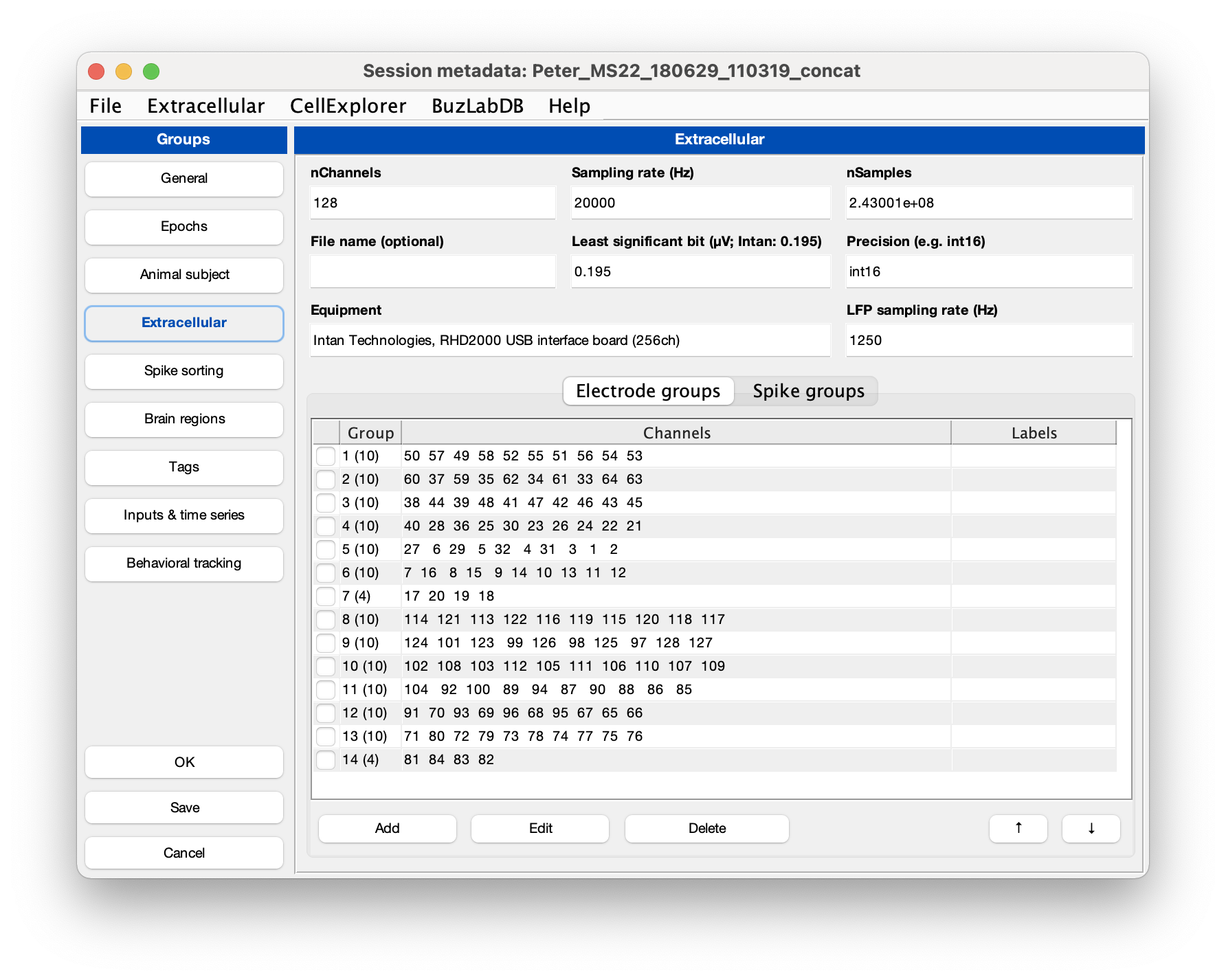
- Run the cell metrics pipeline
ProcessCellMetricsusing the session struct as inputcell_metrics = ProcessCellMetrics('session', session,'showGUI',true);Setting showGUI to true will display the GUI shown below allowing you to validate parameters and settings for
ProcessCellMetrics. You can click the button Validate metadata to show a table with metadata relevant to the processing. Fields requiring your attention will be highlighted in red; optional fields in blue.
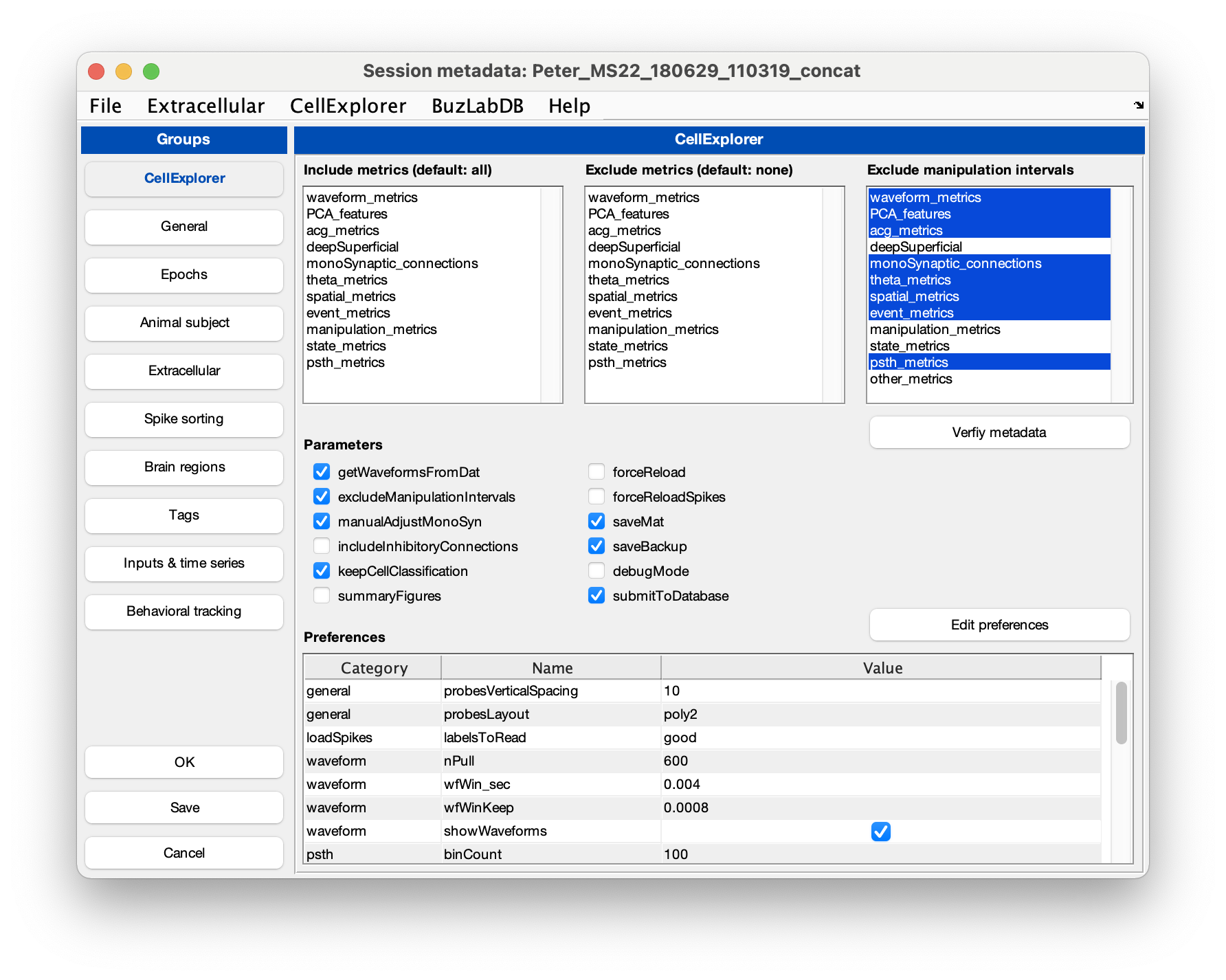
- Visualize the cell metrics in CellExplorer
cell_metrics = CellExplorer('metrics',cell_metrics); - Open multiple session from their basepaths
basenames = {'Rat08-20130708','Rat08-20130709'}; basepaths = {'/Volumes/buzsakilab/Buzsakilabspace/Datasets/GirardeauG/Rat08/Rat08-20130708','/Volumes/buzsakilab/Buzsakilabspace/Datasets/GirardeauG/Rat08/Rat08-20130709'}; cell_metrics = loadCellMetricsBatch('basepaths',basepaths,'basenames',basenames); cell_metrics = CellExplorer('metrics',cell_metrics); - Curate your cells in CellExplorer and save the metrics
- You may use the script
loadCellMetricsfor further analysis using the metrics as filters- Get cells labeled as Interneuron
cell_metrics_idxs1 = loadCellMetrics('cell_metrics',cell_metrics,'putativeCellType',{'Interneuron'}); - Get cells that have the groundTruthClassification tag Axoaxonic
cell_metrics_idxs2 = loadCellMetrics('cell_metrics',cell_metrics,'groundTruthClassification',{'Axoaxonic'});
- Get cells labeled as Interneuron
The cell metrics follows the definition here. E. g. the filtered waveforms are stored in the field cell_metrics.waveforms.filt, and the firing rates in the field: cell_metrics.firingRate.