Neuropixels tutorial
This tutorial shows how to process a Neuropixels dataset. It is based on the online presentation of CellExplorer at the Neuropixels course in 2022. The presentation included an introduction with slides and a demo in Matlab (11 minutes long - the video is embedded below).
This tutorial includes:
- Preparation of the session metadata
- Running the cell metrics pipeline
- Visualizing the cell metrics in CellExplorer
- Using NeuroScope2 to view the raw data and derived data.
The tutorial is also available as a separate Matlab script (tutorials/CellExplorer_NeuropixelsTutorial.m).
1. Generate session metadata struct using the template script and display the metadata in the session gui
For this tutorial, I used a Neuropixels dataset from a pilot study performed on a rat (384 channels, 200GB, ~2.5 hours).
First we define the basepath of the dataset, and go to that folder in Matlab:
basepath = '/Volumes/Peter_SSD_4/NeuropixelsData/PP02/PP02_2020-07-10';
cd(basepath)
The dataset should ideally consist of the raw data basename.dat, and spike sorted data. For this session we have a number of files from various sources (check the video above for further details):
- PP02_2020-07-10.dat : raw data (format define here)
- rez.mat : metadata from KiloSort (format defined on the KiloSort wiki)
- PP02_2020-07-10.xml : metadata from NeuroSuite
- *.npy and *.tsv files : Spike data from Phy (files described on the Phy website)
SpikeGLX and OpenEphys stores the raw data as a bin file and a low-pass filtered LFP file in another folder. The .dat file and the .bin file are the same underlying format but different extensions. You can either rename the .bin file to .dat, or specify the relative path and name of the file in the session metadata:
session.extracellular.fileName = 'rawdata.bin';.This is also true for the LFP file, but here you need to create a copy or rename the lfp file to basename.lfp. Also makre sure to specify the correct sampling rate for the lfp file:
session.extracellular.srLfp= 2500;
Using a template script we will generate and import the session-level metadata from these files. It will import metadata from the rez.mat file, from the Neurosuite xml file and the npy files:
session = sessionTemplate(basepath);
Next, we will use the session-GUI to inspect the generated session struct. You can learn more about the GUI here. The session-GUI is organized in the same way as the underlying session-level metadata struct which is defined here:
session = gui_session(session);
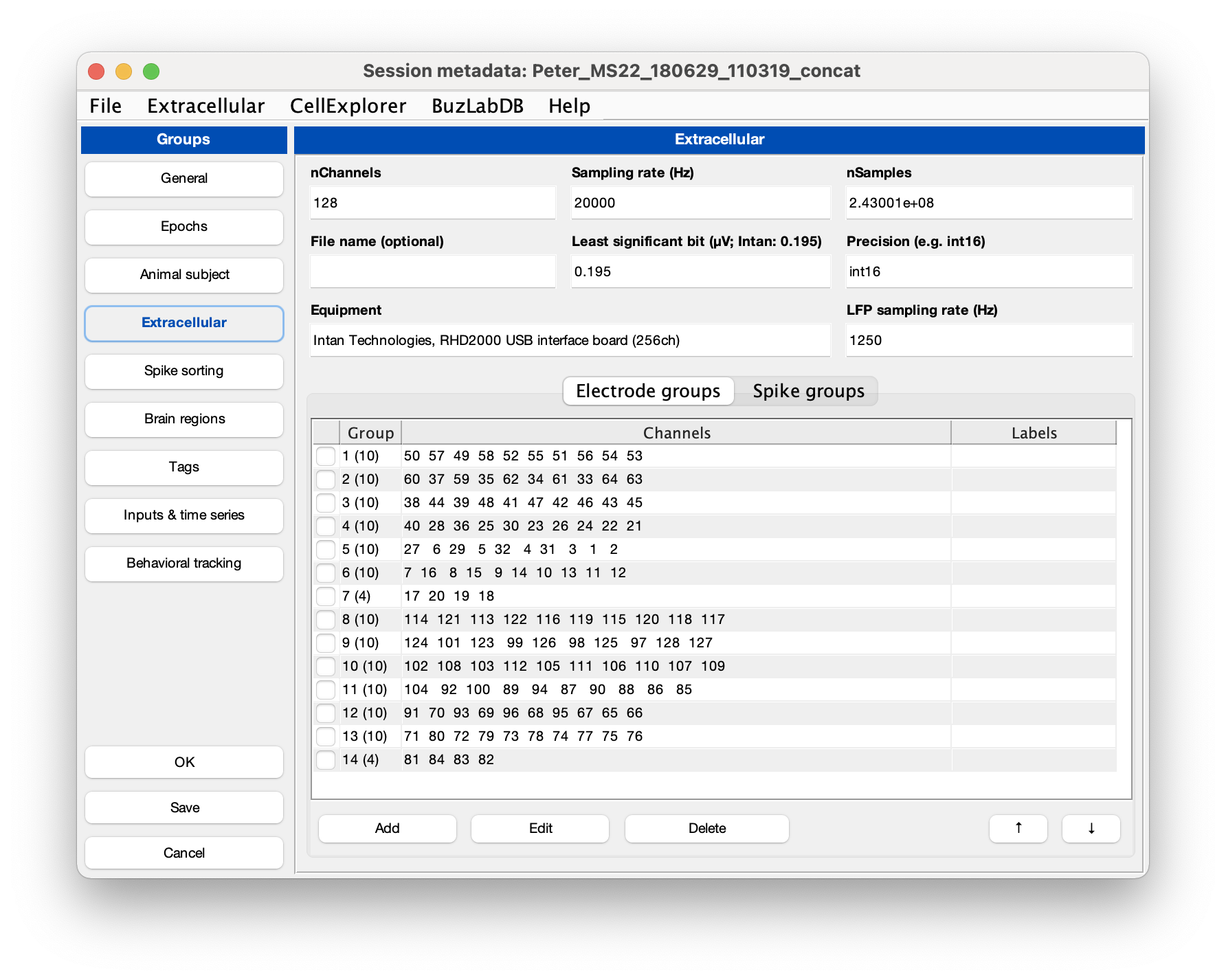
Make sure that the extracellular tab is filled out correctly for your data (see example screenshot below). The template script can extracted existing metadata from a Neuroscope compatible basename.xml, from Intan’s info.rhd file, from KiloSort’s rez.mat file, and from a basename.sessionInfo.mat (Buzcode) file.
Neuropixels recordings typically will have 384 channels, sampled at 30 kHz. The channels are ordered staggered along the probe, starting from the tip of the probe. Least significant bit is 0.195 µV.
There is a script you can use to validate required and optional fields for CellExplorer. It will show you a table with the list of parameters and their values in the session-level metadata struct. It cannot validate if the parameters are correctly entered for you specific data, e.g. it cannot validate your channel count or sampling rate, only if the parameters has been set. Fields requiring your attention will be highlighted in red; optional fields in blue.
validateSessionStruct(session);
2. Run the cell metrics pipeline ProcessCellMetrics using the session struct as input
Now we can run the processing pipeline.
cell_metrics = ProcessCellMetrics('session', session,'excludeMetrics',{'monoSynaptic_connections'},'showWaveforms',false,'sessionSummaryFigure',false,'showGUI',true);
Setting showGUI to true will display the GUI shown below allowing you to validate parameters and settings for ProcessCellMetrics. You can click the button Validate metadata to show the validation table with metadata relevant to the processing.
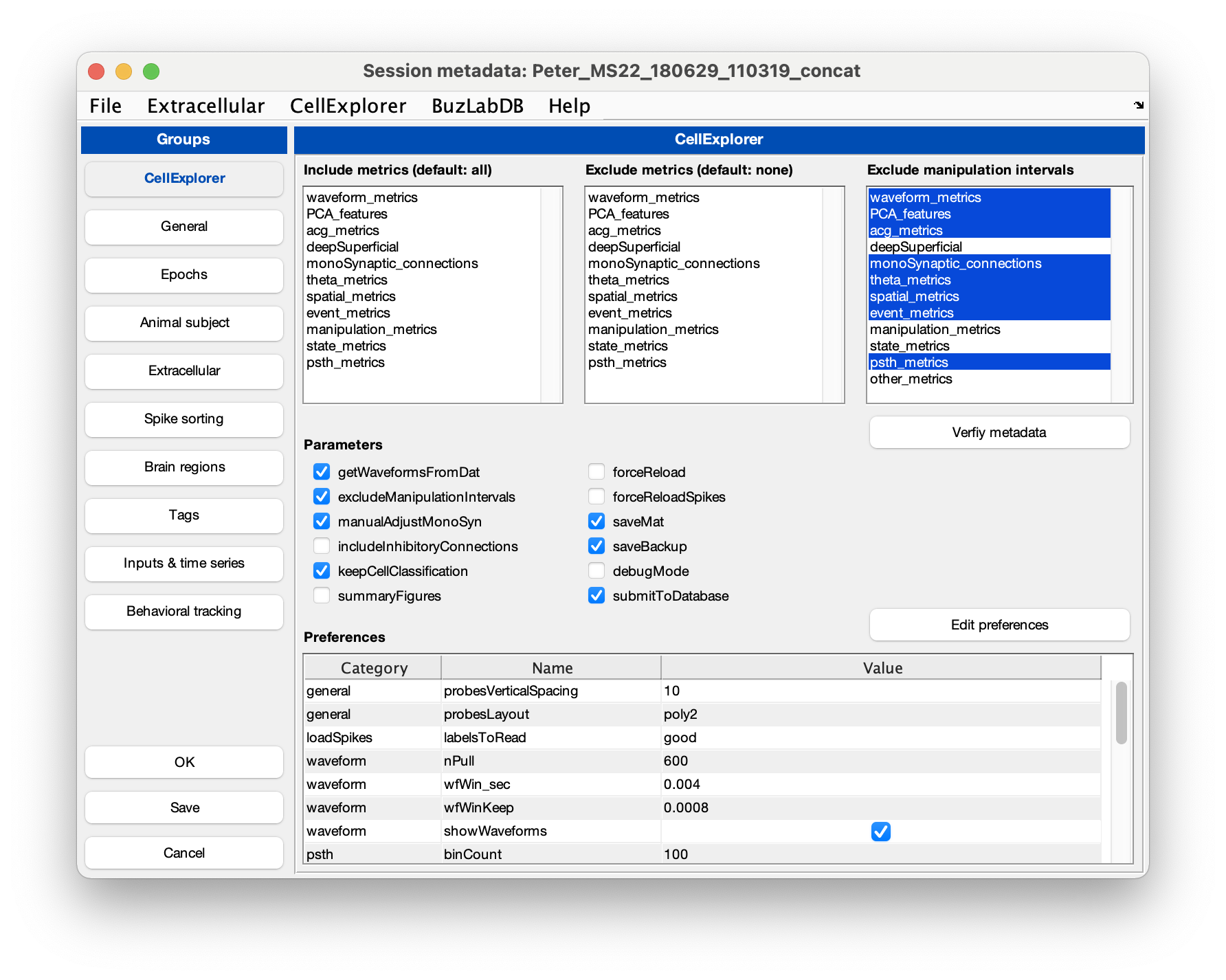
Several files are generated by the pipeline (if they do not already exist):
- basename.cell_metrics.cellinfo.mat : the cell metrics
- basename.session.mat : the session-level metadata
- basename.spikes.cellinfo.mat : the spikes struct
All files and structs are documented here and the cell metrics follows the definition here.
Once created the files can be loaded with dedicated scripts:
session = loadSession; # Loads the session metadata
cell_metrics = loadCellMetrics; # Loads the cell metrics
spikes = loadSpikes; # Loads the spikes.
3.1 Visualize the cell metrics in CellExplorer
Once the metrics has been computed, we can use CellExplorer to visualize them:
cell_metrics = CellExplorer('metrics',cell_metrics);
3.2 Batch of sessions recorded from mice and rats
The power of CellExplorer really shows when working in bath-mode. Here I am loading a pregenerated batch (3686 cells from 111 sessions):
load('/Volumes/Peter_SSD_4/cell_metrics/cell_metrics_peter_viktor.mat');
cell_metrics = CellExplorer('metrics',cell_metrics);
To work in batch-mode you can define the list basepaths and basenames of your sessions, and combine them with a dedicated batch script loadCellMetricsBatch, before running CellExplorer
basepaths = {'/your/data/path/basename1/','/your/data/path/basename2/'};
basenames = {'basename1','basename2'};
cell_metrics = loadCellMetricsBatch('basepaths',basepaths,'basenames',basenames);
cell_metrics = CellExplorer('metrics',cell_metrics);
4. NeuroScope2
Here I am using NeuroScope2 to view a dataset recorded with two 6-shank silicon probes implanted bilaterally in CA1 (128 channels; 150 cells)
cd('/Volumes/Peter_SSD_4/CellExplorerTutorial/MS22/Peter_MS22_180629_110319_concat');
NeuroScope2
Example dataset shown in NeuroScope2 is available to download
Please see the download instructions. The dataset contains a raw dat file, an lfp file, session metadata, spikes, brain states, behavior, events data, and digital and analog traces.
Viewing a Neuropixels recording in NeuroScope2
Matlab’s plot performance scales non-linearly with the number of raw data points, but linearly with the number of channels plotted. For optimized performance, the windows duration should be kept below 0.6 sec when viewing the raw data for a Neuropixels recording with 384 channels sampled at 30kHz. Neuropixels recordings has a DC offset that is recommended to remove when viewing the traces by selecting “Remove DC from ephys traces” from the settings menu in NeuroScope2.